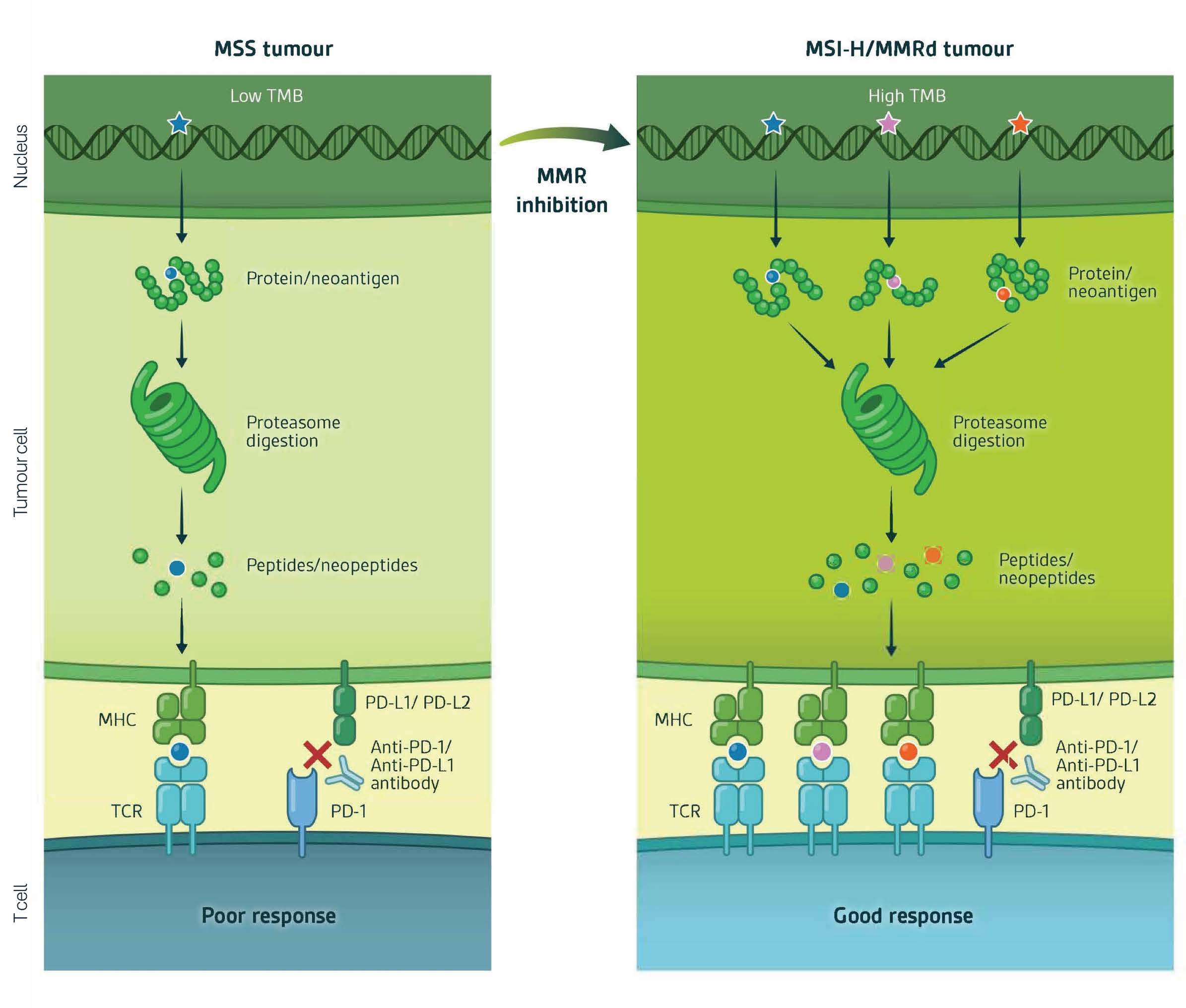
Components of DNA MMR are genetically lost or mutated in up to 20% of certain patient tumours, notably colorectal, endometrial, rectal, and gastric cancers. MMR-deficient cancers are commonly characterised by the accumulation of DNA mutations during replication. These accumulated mutations comprise both single nucleotide variants (SNVs) and indels/microsatellites. Accumulation of DNA SNVs and Indels over time engenders an immune responsive state. (Figure 2).
Accumulation of gained SNVs and indels is quantified by Tumour Mutational Burden (TMB) whilst the origin of the gained mutations is determined by the fingerprint signature of the gained mutational patterns which are characteristic of MMR-deficiency (Nature click here and Nucleic Acids click here ).